By: Martin Wang, Marcelo Kuroda
The objective of my research project was to determine the viability of molybdenum disulfide (MoS2) in low-power electronic devices and flexible optoelectronics. The motivation for this research topic lies in its ramifications for a crucial and ubiquitous electronic component: the transistor. Traditionally, transistors have relied on the physical properties of their bulk materials; however, as modern circuit components continue to scale down, materials with reduced dimensionality have emerged as alternative solutions for optoelectronic applications. This change in paradigm follows the ne w challenges posed by modern technological applications.
Among the novel alternative materials, MoS2 has several remarkable properties that make it interesting in the context of transistor technology. First, MoS2 has the property of being a two-dimensional material– it exists naturally in stable, few-atom-wide layers. The advantage of 2D materials is twofold: it lacks the unstable hanging bonds which cause the shortcomings traditional bulk materials such as silicon, and it enables more flexible electronics by allowing different layers to slide by one an other. The second interesting property of MoS2 is its two phases: the 1T phase and the 2H phase. These two phases of MoS2 use the same atoms in different molecular geometries, producing different electrical properties– the 1T phase is a metallic conductor while the 2H phase is a semiconductor. Thus MoS2 can, by itself, singly form both the transistor contacts and channel. However, this sy stem comes with its own difficult challenges to overcome: when MoS2 is interfaced with bulk metals, it suffers from Fermi-level pinning that results in a large contact resistance.
To probe the electronic properties of MoS2 , we employed Density Functional Theory (DFT) within the generalized gradient approximation (GGA) using the pseudo-potential method. The MoS2 band structure calculations for several vertical heterostructures were conducted using the computing resources provided by the Alabama Supercomputing Center, as well as Auburn’s own CASIC cluster. These calculations aimed to establish the possibility of circumventing the problem of Fermi-level pinning using MoS2 heterostructures.
The analysis began with relaxation calculations which were used to determine the proper lattice constant of the MoS2 crystal models and the relative positions of the MoS2 atoms. The project then proceeded to calculate the electron bands for MoS2 heterostructures ranging from the two layer 1T-2H to the four layer 1T-2H-2H-2H. Finally, the calculations applied field and charge to the models and generated the band st ructures that could be analyzed to determine the electric susceptibility.
The analysis of band movement with respect to layer number and configuration revealed that MoS2 produced relatively large electric susceptibility when the 1T phase is placed above the 2H phases. This susceptibility increased approximately linearly with additional 2H layers. This property alleviates the Fermi level pinning and offers promising indications for the viability of MoS2 in low-power electronic devices and flexible optoelectronics. Moving forward, more sophisticated and computationally expensive calculations must to be performed to overcome the limitations of Density Functional Theory and further validate our findings.
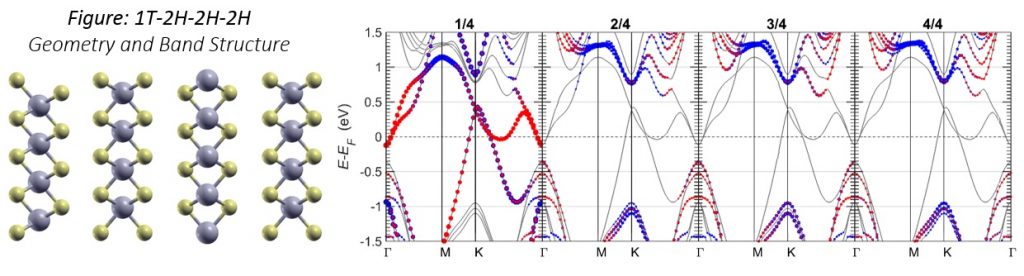
On the left, the atomic model of the 1T-2H-2H-2H heterostructure is shown. Yellow atoms asulfur, and blue atoms are molybdenum. The 1T phase MoS2 has unaligned sulfur atoms whereas the 2H phase MoS2 has aligned sulfur atoms. On the right, the band structure of 1T-2H-2H-2H is shown: the energy of the band with respect to reciprocal space. Γ, M, and K are points of interest on the closed loop. Blue dots represent molybdenum contributions, and red dots represent sulfur contributions; dot size scales with magnitude of contribution. Each subgraph (labeled N/4) cor responds to a different layer in the
1T-2H-2H-2H heterostructure.
Statement of Research Advisor
Martin Wang has performed a large number of first principles calculations and created computational tools to analyze the outcomes of those simulations. These encouraging results show that the behavior in MoS2 heterostructures is remarkably different from those using bulk metals, opening new possibilities for MoS2-based electronic applications.
—Marcelo Kuroda, Physics
Last Modified: